

I have been having a discussion on another forum, on how it is possible for water to pickup chlorophyll in an extraction, when chlorophyll is basically a hydrocarbon, which is mostly insoluble in water.
I did enough research to know that I was in over my head with chemistry that I took on the late fifties and early sixties, so I asked Joe, our budding biochemist, to take a run at it.
In quick summary, before Joe's response, "define soluble?" The word soluble means different things in biochemistry, than it does in organic chemistry, because of the behavior of the molecules of life.
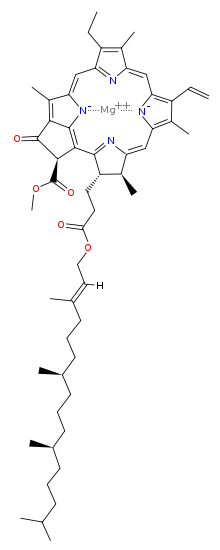
The chlorophyll molecule has a magnesium (Mg) at its rings center, which makes it ionic and water-loving (hydrophilic) and a ring that is water fearing (hydrophobic) with carbonyl groups near a tail that make it polar (also hydrophilic).
It is held in place in the plant within a water-soluble material known as water-soluble chlorophyll-binding protein (WSCP). WSCP is soluble in water, and mostly insoluble in polar alkane alcohols and non polar alkanes.
Chlorophyll is readily soluble in alcohol, mostly insoluble in non polar alkanes like butane and hexane, and has some special relationships with polar water, because of its polar and ionic groups.
Mostly is a key word in all cases, because of chlorophylls charged polar end and non polar hydrocarbon ring with the ionic Mg.
Before I finish summarizing, here is Joe's response to the question of how water can remove and transport basically insoluble chlorophyll molecules, as well as how saturating water with NaCl table salt aids in the process of washing unwanted chlorophyll out of extractions that have gone awry:
Chlorophyll Info by Joe
Chlorophyll is an intra-membrane chemical within a thylakoid. A thylakoid is a membrane-bound compartment inside chloroplasts.  The thylakoid membranes of higher plants are composed primarily of phospholipids and galactolipids that are asymmetrically arranged along and across the membranes.
The thylakoid membranes of higher plants are composed primarily of phospholipids and galactolipids that are asymmetrically arranged along and across the membranes. 
Chlorophyll is shown as photosystem I and II in this illustration.
Both phospholipids and galatolipids have hydrophilic (water-loving) heads and hydrophobic (water fearing) tails.
In biology this theme is used in almost all life forms to compartmentalized for energy storage, isolate invaders or encase their genome to protect it and many others reasons.
Solubility is a term that has more than one definition. In inorganic chemistry it refers to waters (or another compound) ability to break covalent and ionic bonds of most compounds, dependent on time temperature and pH. The “solublized” atoms are then bonded to their polar opposite ion H3 (+) or OH (-) and are in solution.
In biology however, it is used in the first tense but, it is also used to describe the ability of an organic molecule or complex to form an association with water and be in solution but not be “solubilized” by it. Proteins and other organic molecules use charged ions such as phosphate (PO4) and Sodium (Na) to form micelles. Micelles are little balls of hydrophobic molecules surrounded by a charged ion.
Just like a cell's membrane bilayer. Sometimes micelles are formed by complexes of proteins surrounding a small molecule for transport through water.
Chlorophyll specifically, is only able to form complexes with other molecules to stay in solution at biological pH (7.4). Its natural environment is at a pH of around 4 not 7.4. At this pH it has a net charge of -2 so that it can form a chemo-gradient for electron transport during photosynthesis.
So since pH =-log [OH-/ H+] when at pH 4 the [H+] concentration is higher than the [OH-] thus creating an environment that is more likely to associate with the (-) charged area of the chlorophyll molecule. Hence the low solubility of unbound chlorophyll in water, the large hydrophobic areas compress together and present their hydrophilic areas to exclude water from the center, becoming a mass that will sediment in water.
The point of this is to illustrate that while purified chlorophyll is not likely to stay in solution in pure water; we don’t extract pure chlorophyll and we don’t use pure water.
We use brine to keep the charge on the phospholipid bilayer (Na+ with PO4-3) and no detergents. The alcohol (ROH+) wants nothing to do with Na+ while in its protonated (H+) state.
Non polar solvents for obvious reasons won’t either and also won’t form much of an emulsion with alcohols in their bent state because; the alcohol is denser and forms a micelles like layer to protect itself from the charged Na+. If there is an excess of alcohol it will start forming an emulsion layer at the upper interface.
The phospholipid bilayer of the chloroplasts and of the thylakoid being intact or mostly so, prevent the chlorophyll from being disassociated with the Na+ water and are able to be excluded from alcohol or non-polar solvents .
Alcohol is able to associate with chlorophyll and proteins in its native conformation but not when bent by Na+ because the electron pool concentrated at the (O-) repels the (-) region of the chlorophyll molecule and the PO4-3 of the membrane bilayer.
If the membranes have been broken up by a detergent or broken down by enzymes then the only way to exclude chlorophyll from a non-polar solvent or alcohol is with lots of Na+ and water. Because of the large non polar area of the chlorophyll molecule it can more easily form a hydrophobic interface and shield the charge in the center.
The salt exposes the charge (because ionic is a stronger bond than Van Der Wals forces) and precipitates the chlorophyll into water.
So in conclusion, chlorophyll is not wholly soluble in water, but in its biological complex is able to associate with it.
By manipulating charge/charge interactions chlorophyll can be forced into solution with water and away from polar organic and non-polar solvents. While it doesn’t meet the inorganic chemistry definition of solubility, it will form micelles complexes with an ionic solution and it can be precipitated from that solution under the right conditions.
From a biological perspective chlorophyll can also be solubilized by any solvent under the right conditions.
Sooooo, back to my summary, it appears that some solvents can wash away the cement binding the chlorophyll in the plant cells and free the chlorophyll to be washed away as a micelles, but it also exposes the chlorophyll to the solvent, which in the case of alcohol, will readily dissolve it and hold it in solution.
Freezing the material prior to an extraction, may be holding both the chlorophyll and the WSCP locked up in ice, so that neither the water present or the solvent can reach the chlorophyll.
Merck Index lists chlorophyll as practically insoluble in non polar solvents, so the difference between practically and totally insoluble may offer a clue, as well as butanes practically insolubility in water.
While most sources list n-Butane as insoluble in water, its actual solubility is 0.0325 vol/vol, at 1 atmosphere pressure and 20C/68F. That is 32 ml/liter, which may be enough to account for the light electric green hue that occurs, by both washing away the WSCP and holding some of the chlorophyll in suspension.
When we saturate the water with salt, before washing a polar extract suspended in a non polar solvent like hexane, it forces the chlorophyll into solution and washes it away.
Explanation by Joe, summarized by Graywolf


SkunkPharm wrote: "Depends on the type of alcohol. I use a three minute soak for frozen material, using chilled ethanol and 30 seconds for chilled Isopropyl." How long would you soak for chilled (-40'C to -50'C) absolute methanol?
No way am I reading all of that. But I ended up here because I was trying to discover how efficiently uv removes the chlorophyll from your trim/bud treated with ipa for an extraction not seeing that after skimming the above but if you can come up with that I'd bet money you can answer my question with ease. If you don't mind that is.. if I had soaked triche covered trim/sugar leaves in isopropyl alcohol and the solution turned green and after leaving it to sit in the sun it's basically dark Amber, if I left it out for longer would this make the oil impotent having the thc destroyed by uv if it has went from completely non see through dark green to Amber? Ik thc is destroyed at a much slower rate than chlorophyll but does it make a substantial difference if I left it in not so bright sunlight for 3 days until it turns much more Amber? I also did this before and noticed a sediment was collecting at the bottom sort of white to golden blonde kind of resembling pure kief and when I dried it out on Pyrex it formed beside the oil, I smoked it in a bong but I didn't notice that scuffy resinous feel on the back of my throat or taste it's terpenes all that great I did feel something from it however, also it wasn't even enough for a bowl on its own but I put it in itself regardless to taste, is that purer, sediment from chlorophyll or dead thc? Is it possible to let the alcohol soak into the bud and shake vigorously then completely allow the chlorophyll to be removed via uv light and still have a strong oils?
UV doesn't remove the chlorophyll, it just breaks it down into its components, which are amber instead of green. Some of those components and some plant waxes may have precipitated out. Isopropanol is a polar solvent, but not as polar as ethanol or methanol, so extracts more non polar plant waxes. Better to avoid extracting them in the first place, by extracting at subzero temperatures. Check out QWISO and QWET: https://skunkpharmresearch.com/qwiso/ https://skunkpharmresearch.com/qwet-extraction/ UV also destroys all of the terpenes, including the phenolic diterpenoid cannabinoids, but not in a flash or they couldn't survive on the plant in the sunshine. I do however suggest developing a process that minimizes using UV to bleach chlorophyll, for the least terpene/terpenoid damage and greatest aromatics/flavor/potency. GW
Respectfully, I do not understand why you mention chlorophyll is a hydrocarbon? It has oxygen in the molecule I believe. A hydrocarbon only has hydrogen and carbon, hence the name. Terps are hydrocarbons, specifically volitile organic hydrocarbons (VOC). This is why when you burn the material to ingest, as in a bong or joint, that even really heavily terp strains are usable. Most of the VOCs present are combustable easily with a flame, but I would say THC is not because 1) if it did combust we would not get high and 2) it is not a VOC or even a hydrocarbon. I use activate carbon in my oil pot adapter on my dab rig. Pellets of it, pure AC. I use a torch to "activate" the thc in it whic provides an extremely smooth vape. The VOCs burn off and the AC holds anto the high temp VOCs very well. There is a,chemical way to,easily remove anything more polar than not - select a highly non-polar solvent, disolve extract in it, then select a solvent more polar but in between the first and water. Add and mix. Now add water slowly to the mix. Layer on top is most non polar and bottom two have the rest, including the green stuff. I use a seperatory funnel then and use a fan to evap the solvent on top. Then a few boils in a hihhly polar solvent, H2O, and into the vac chamber. It will take a lot of the terps yu get in first run short path distillation with it as well. This makes me wanna vape. Thank you for the sight SkunkPharm!
I just got a short path can you give me a quick summary of your separation process of extracts of cbd/thc. I am working with a dark green and waxy material.
The short path apparatus design can be a huge factor in how it works. I habe seen short vids online of others demonstrating short path and their equipement is horsepower heavy and involves industrial size chillers. My set is a small honby set and my vacuum pump is a large dual stage unit, but regulating the vacuum at a set reading is tough so I do use the vacuum to help but I do not "pull" all the thc out as in the big rigs with the big pumps, and I am not concerned one bit about how hot my extract gets. You did not ask about h2O co-distillation which I have done many times with huge success. I work with 28 grams or less of raw extract doing this. Anything left in a distillation that sticks to the inside of the device is called "hold up". At low quatities the hold up is a big percentage. Easily recovered but just letting you know. Irregardless of vacuum used, the challenge is to keep the short path hotter than 315 F all.the way until the glassware turns down towards the receptical vessel. I have a heating mantel for the boiling vessel. I wrap the exposed glass in fiberglass matt loosley like a blanket. Two loose layers covering the entire aparatus all the way up and turn the corner into the condenser. I work by my kitchen sink. Always wok near a sink that you can push the enitre apparatus into in an emergency. Things are gonna get very hot. Secure the fiverglass matt (available on Amazon in hobby section very cheap) wih a metal stick pin. Then I cut up some old wool dog sweaters and draped them over the fiberglass. The glass inside MUST be able to hold the heat above 315 F ALL the way up until the condenser or it will simply reflux back into the boiling vessel. Since I have a kitchen sink next to it and my condeser is run off of the tap then instead of cold water in the condensed I run hot top water into it. Hot tap water is still below the condensation point of thc and a hot condenser greatly reduces hold up. The hot water helps keep the upper portion of the apparatus hot enough to keep the thc in vapor form until it heads for the collection vessel. I use a fan to cool the recivers and the "cow". The first part of the fraction is clear. THC is light yellow. The clear is likely many things but predominently pinene at distillation temps smells absolutely vile. Vomit city. The pinene of the first fraction will taint the taste of thc if nothing further is done after the run. Since I use such tiny quaritities a second run to remove the residual pinene is not viable for me so I remove the pinene with a flame process and isopropyl alcohol in similar manner to how a chef removes unwanted flavors from food using a technique known as "flambe". This site has asked me to refrain from posting videos of this however there are thousands of videos demonstrating the flambe technique on any number of cooking sites. Check these out if you seek a quick way to remove the residual pinene. Pinene is highly flamable at room temperature - thc is not flamable. The flambe process safely ignites the volitile pinene thus removing it. Please follow all safety procedures comtained in the cookbooks for flambe cooking. I have attempted to use activated carbon to trap the pinene and it does work, however, careful weighing of products reveal that extract is absorbed by weight on a one to one ratio with activated carbon. One gram of carbon pulls out one gram of extract. Carbon pellets smoke just fine in a pipe and produce the sweetest smoke there is. I even dab onto carbon pellets and smoke off of them an enjoy it greatly, but it is not relavent to this question. Co distilling the extract in h2O is infinately easier and is carried out at around 98F, but the considerations would take a seperate section to demonstrate. I have seceral videos of how I did it and the h2O process has the added benefit of helping remove the pinene because pinene is partially miscible in water. I use exactly the same rig as short path except for a seperatory funnel for the h2O feed into the boiling flask. It takes volumes of water to carry this out. Here is what water co distillation looks like. https://vimeo.com/180067017
Is the h2o codistillation written up anywhere on how to setup and run? Thanks.
[…] Chlorophyll Pickup in Extractions | Skunk Pharm Research … – I have been having a discussion on another forum, on how it is possible for water to pickup chlorophyll in an extraction, when chlorophyll is basically a hydrocarbon … […]
Is it possible to convert the wheatgrass juice into a vapor with added flavors without loss of efficacy in the process?
Removing terps is a good idea. Try boiling your extract and take a good whiff of the first vapors,that begin to rise at about 280 F - 300 F. It is absolutely repugnant. When terps are not in marijuanna products, people call them industrial solvents. I believe a good solvent helps the med penetrate the mucous layer in the lungs thereby delivering the med quicker, but ling term inhalation of industrial solvents causes me to have a constant hacking cough. So i remove a good portion and in the process the vapor becomes sweet and fruity.
Hi. I was wondering if you could tell me of cheap substance to remove chlorophyll. As in attack chlorophyll and wipe it out.
sooo i think i stumble on to a way of removing the unwanted from 190 proof green dragon ......this would be for edibles only as i am using a non smokable oil in this method .........but before i open my self to ridicule does any one have access to lab
Hi there. Great scientific explanation. I barely remember all this from school. Brings back memories. Anyway, my question is simple. All I'm trying to do, is to be able to turn the plant material into e-cig juice for vaporizing. I am on the road too much and e-cig juice is the most convenient. I've tried the quick wash with everclear. Turned into a dark oil, which i eventually dissolved in PG, VG PEG mixture and there's my e-juice. A purer e-juice might taste better, not like grass in your mouth and the smell of the vapor. Reading all of your articles, could I, instead of doing a quick wash in ethanol (everclear), do a long wash. Like extract the hell out of everything from the plant material. Soak it for weeks or whatever. Then after the first strain, I can throw away the left over plant material knowing that i've got all (theoretically) of the stuff, good and bad in the ethanol now. Can I then just rinse the ethanol in a brine solution over and over and over until all the waxes, lipids etc... are gone? (I'm over generalizing here with my choice of words...... but you know what I mean... i hope)
Freeze the ethanol and plant material in the freezer for several hours or pack the plant material and ethanol in a ice chest with dry ice (gets colder than a freezer). Then do the extraction and try to limit the exposure of the plant material to the solvent by packing a filter basket full of the plant material and pouring the ethanol over it as quickly as possible instead of combining them both in a jar. I can usually get 3 or 4 pours reusing the same ethanol before it starts getting dark. Depending on how refined you want it you can then filter it though a coffee filter, or use a separatory funnel to eliminate more contaminants like fats and waxes. With this method you should get an amber oil instead of green. Evaporate the ethanol in a pyrex dish on low heat in a hot water bath (under 120*f) I wouldn't use fresh trim or buds because the ethanol will pull the water and more of the sugars out. I am an organic freak so i always used ethanol, but I have tried iso and iso isn't as harsh of a solvent as ethanol. i have found that when both are frozen i can expose the plant material to the iso for a longer time before the chlorophyll begins to break down.
Hey Kenny did u ever try using that brine solution and if u did how did it turn out? Thanks
Hi Cruz, no, actually I let this go on the wayside. Life happened. Anyway, now that you asked. I think I will try it. I'll do a small batch and let you all know if the brining does anything.....
Will the became stay in the solvent? How do you desperate brine and hexane? The became from solvent? Just trying to make our dark run bho golden...
Or just use fresh frozen trim or buds!
Okay. Here's a CRAZY idea. (BUT, it's JUST an idea- No STORY-BOARD-) We ALL hate having Chlorophil in our Soluables, but when using ISO, we always get Green along with the good stuff in our solution. RIGHT? Well, since it dissolves in H20 and the GOOD STUFF does NOT, lets SOAK our Leafy Green stuff in plain old Cold Water. We aren't going to shake it or move it around, and we put a Tablespoon Per quart Per 14 Grams OVERNIGHT. In the Morning, the Ice-Cold Water will be BRIGHT GREEN. Disgard it after GENTLY straining out the Cold Buds and Leaf. Dry completely. COMPLETELY. "TA-DA!" WAY LESS 'soluble JUNK to START WITH = Less SURFACE Chlorophyll to come-off during a 30-second 'Bath' later. Nice and YELLOW to Amber Liquid means smooth! (Someone could soak an EXTRA night if desired-)
The soak in cold water will also remove all terpenes, you will get less chlorophyll in your concentrate but you will also lose all terpinoids, which unless you are using a cold extraction method and a vaccum to purge your oil if you use ethanol or any alcohol for that matter you are going to lose terps, I suggest a -100 degree N Butane saturation for 45 minutes, if not in a closed loop then use a mason jar and seal it and put it in the freezer or dry ice bath, the butane will not break the jar, do not fill the jar more than halfway full as the vapor bubble contains the terpenes that are most volatile, ones you want to keep, anymore questions feel free to email me at dabdad1984@gmail.com
I had a leaf wash w/ IPA that was just filthy,I took my test tubes and filled half with DI and slowly filled the tube to the top, after about a week a precipitate collected on the bottom so I repeated this procedure w/ a volumetric flask filling the bulb w/ DI and the stem w/ 'dirty' solution w/ the same results, I will need to repeat the procedure in order to collect enough to test, since I have no analytical equip. or cash for any reagents any idea what I'm collectting?... OH, almost forgot I repeated this procedure in the tube with graphite electrodes wired to a usb power cube so approx. 5volts w/ max 500mA, the solution near the electrodes grew a lighter yellowish color(the chloro...) but I also had a control tube and collected about a 3rd more preccipitate from the electolysis. I didn't know the correct posting for this so I hope hears fine. Like everyone else I want to say great work, and boy this feels nice after years of clandestine shadows! thanks, DF
[…] tobacco but probably applicable to other herbs as well. Ooh.. that's not my understanding. From Chlorophyll Pickup in Extractions.. ..Chlorophyll is readily soluble in alcohol, mostly insoluble in non polar alkanes like […]
Has anyone run test to show what cannabinoids / terpenes are removed by activated carbon and at what percent of final quantity? We would like to know the effects (loss of total return) of this filtering on iso wash and whether prior decarboxylation (of extracted oil and/or plant materials) has an effect on the return of active cannabinoids in final products. Love your work guys! so nice to see this quality of info on the web with so much junk science and hearsay clouding up the scene.
I'm trying to keep my oil extraction process as simple/healthy as possible... Have you ever extracted using 190 proof ethanol below -40 F degrees? Would this lock up water solubles into ice and provide extra wash time (assuming temps stay below -40) to really get a nice extraction using food grade alcohol? I am considering purchasing a cryo-freezer to perform the extraction, for steady temp control. Both the wash and strain inside the freezer and then using an essential oil still to reclaim solvent and hopefully save some terpenes in the process. Wouldn't using ethanol in this fashion help me avoid winterization and leave me with a nice clean oil? I'm also looking at Cascade Tek's TVO-2 for final purge. All oil made in this manner is intended to fight cancer and promote a healthier ECS. Thanks for all you do!
We haven't done a QWET that cold, but it certainly would tie up the water and water solubles. I suspect you can do a fine job warmer than that, but it is the right direction to be experimenting in. You could mock it up using a dry ice and denatured alcohol bath to chill a sample and see how long you can soak it without picking up non targeted components, as well as what the lower temperature does to extraction rates. TVO-2 is a nice oven. After using one in a joint R&D project with Cascade TEK, we bought one of our own.
Thanks much for getting back, If I might ask... what would be the benefits of using denatured over undenatured? I was hoping to use everclear or something organic in that proof to keep the process down to 1 solvent. A gentlemen out of Yelm, Washington has been working with grape ethanol and low extraction temps for a little while now. He has of lately been posting results from Integrity Labs out of Olympia, WA with VERY high scores. The color and test results kinda have my head spinning right now. I'm interested in achieving these scores along with preserving terpenes through relfuxing/decarbing the end product in my still...Should make a nice healthy little product to ward off those nasty inflammatory cytokines. https://www.flickr.com/photos/54396803@N05/15238614730/in/photostream/ https://www.flickr.com/photos/54396803@N05/15238798000/in/photostream/ Thank you for your time and experience
Sorry about the giant bulletin board on your blog...didn't expect those pasted links in my last post to blow up in here.
We use 190 proof Everclear, which we get at the liquor store. 190 proof/95.6% EtOH, extracts the same whether from beer or wine. Fruit has pectin, which produces more methanol than grains, but that is ostensibly removed in the heads. The only reason I can see to use denatured, besides less money, is if that is your only option. It adds the extra burden of needing greater assurance of an adequate purge.
Great! I will be ordering a freezer within the week. Dry Ice as well as liquid Nitro were both successful trials. The freezer should pay itself off in the long run, as the price of ice would add up quickly over time. Again, Thank You!
Good job on the trials! Bon appetite!
I would like to know the answer to this question as well please, creating above said product ^^ ? Thanks for everything you have done
I have often used 190 ever clear at -40 and as long as you wash it quickly without letting it soak into the material then you can get the most golden colored qwet but if you negatively charge your ever clear with pink Himalayan salt you will be able to let it sit longer and as a added benefit it will have a pH over 7.0 which is added benefit with cancer fighting products
Hey Capn, can you provide more details on charging ethanol with salt? How do you remove the salt? How much salt? Thx.
If you boil the alcohol with stems in it with proper ventilation of course, it won't take more then 1/2 hr. If you choose to macerate the stems you will wait a week. Heat is the way to go!
I have a 2 gallon stainless steel bucket. I'm planning to make stamina I have plenty of stems I plan on putting alcohol in the bucket. How long do I soak the strong how many stamps do I have to soak to make something viable. Thanks for any info.
Depends on the type of alcohol. I use a three minute soak for frozen material, using chilled ethanol and 30 seconds for chilled Isopropyl.
Define something viable? What would you like to end up with as a product?
Hey Skunk pharm guys, I've been playing around for a while,but not really satisfied yet ! Like you said,some people need a more refined end product. That is what I strive for. I would really like to get together in person with some one,and may be come up with one method that I can count on every time ! Hope to hear from you soon. Thanks Ron
I've wanted to do some experamenting with phosphoric acid to remove chlorophyl for a while. I read on some pages about vegetable oil processing that they heat vegetable to higher than the bp of water and agitate it with phosphoric acid and the phosphoric reacts with the clorophyll and produces insoluble compounds that arent dissolved in the oil and will never be able to mix with the oil again. the precipitates can be filtered out. The process appearantly is very effective. they report up to 99 percent reduction in clorophyll, and more phosphoric acid makes it even more effective. So if we weren't a factory trying to conserve phosphoric acid it would be even more effective. also, veg oil refineries use alkali refining to purify oil and remove fatty acids. this wouldn't work with normal mj extract because it would remove all the thc-acid, but if you decarboxylated first, I believe you could wash the oil with a NaOH solution and get rid of much color! They say that all the colorfull compounds get removed with alkali refining. How about we take fresh material and pack it in an essential oil still and get all those turpenes and shit, and then dry out the plant material, extract with some solvent, polar or non-polar or whatever, and then do some processing. the decarboxylated step could be the same as the phosphoric acid wash. put the extracted oil in a metal container and add phosphoric acid. put the metal container in a hot oil bath for decarboxylating, at a good reasonable temp for a short efficient decarb, and stir and agitate to mix the phosphoric acid. cool, then add non polar solvent, and get that decarbed, de-clorophylled oil in a solution and put in a flask and wash with a sodium hydrixide solution to get all the remaining red color and fatty acids. then you have that beautiful decarbed clorophyll free crystal clear hexane layer, evaporate that, and add some ethyl, and do the winterizing step.. at the end, try and mix in those essential oils you distilled in the beginning with the beautiful decarbed oil. then you have the dankest of the dank. I wanna do this, I'll let you know how it goes.
How did that go?
How did that go ?
I have done exactly this process! It works! However I dont "re-fortify" said oil w essential oil extracted first I use the denatured hash oil for edibles thus making the edibles devoid of the green and Ganga taste so many peeps have grown accustomed to when making stoner food. I say this method is more adaptable as a medicine for all undesirable properties are removed before the oil is decarbed and blended with my food stuffs. Also now I have incredibly wonderful essential oils of the varieties I grow to use elsewere in other products we also produce... Highly proprietary and the first I've mentioned it publicaly as well. I have another yield increase technique that I have yet seen published but don't want to be called a charlatan for my claim that it is approx 2.5 times greater then the 2.0 grams per watt that is the current published max yield of any system for cannabis... Yea I know too good to be true! So I'll keep it to myself... Maybe I could be talked out of this info by the right person.
I've scene the new light systems. I am reluctant to buy since things change fast in this industy
I have been getting good results using NaOH and raw CO2 extract. Basically I treat the raw oil as fats for making soap. Fatty acids and triglyceride oils are responsible for making waxes so hard to separate through Winterizing. By saponifying the fatty acids and oils, they are rendered water soluble. Terpenes and cannabinoids are not saponified, BTW. The waxes are no longer tied to the oils and will float to the surface, the oils are now soaps dissolved in water, and the 'medicine' will gently float to the bottom. I have processed Winterized oils and removed 20-40% of the starting mass. The fun part is the amount of color this gets out of the raw oils. And I like the 'shatter' that I get from CO2!!!!! I am intrigued by the phosphoric acid/brine step.
Interesting thought! Thanks for sharing! How is monoterpene retention? What percent NaOH solution, temperature, and ratios? What'ca doing with the soap?
Hey! I followed some on-line 'lye calculators' for home-made soap. I assumed that 1/3 of the extract would be 'junk oils' and that the S.P. value would be the same as Hemp Seed oil. Mixed up the hydroxide solution to about 20%. Got to leave it wet! [One mole of triglyceride needs three alkali ions and three water molecules to make soap] The 'medicine' is not affected by low temp saponification of the fatty acids. Soaping does seem to work better when the oil is fully decarboxylated. It is almost magical, watching the wax drift up to the surface and the goodies sink to the bottom. Oh, did I mention that after making soap it is dissolved in about 4x volume of luke-warm water? I am currently washing the soap down the drain [boo!] as it has WAY too much residual alkali. I am over saponifying the mixture to make sure to get all the oils out. As for Terpenes, it is hard to say as the source material was a discard. That being said, there was a batch or two that smelled just like fresh green herb.
Hey Greg, I was just wondering if you could lay your steps from beginning to end of the saponification process. My last/first run with this method resulted in an oatmeal textured final product and no separation at all. The soap recipe: - 2 ounces of hemp seed oil - 7.61 grams of lye (.1359 ounce/ounce of oil) - 18.66 grams of water (1/3 of oil amount in ounces) - Used QWISO extract and melted into heated hemp seed oil Process: 1. Mixed lye into water and stirred until all dissolved. 2. Once dissolved, mix lye and water with hemp seed oil. 3. Heated mixture to 90°C and hand stirred. 4. After about 10 minutes the bottom of the mixture started to harden. Began mixing this solidified bottom which turned the rest of the mixture into an oatmeal like paste. 5. Removed from the heat to see if separation would occur, but had to throw out the entire mixture due to everything being turned into just a brick of soap basically. Did you use heat for your process or did you just use a cold soap recipe and stir until the mixture began separating by itself (waxy bits floating to the surface while the medicine sinks to the bottom). If you have any tips or instructions on this process it would be greatly appreciated. Best regards, Aaron
Great article! I really appreciate you guys maintaining such an educated and educating site. KEEP IT UP :-D
A friend of mine uses charcoal and then strains it out after his alcohol extraction, the oil is then a cherry red color? What is this from? Is it safe? Is there any way to get rid of this? Thank you I really like all your information on your site!
I believe the red stuff is the broken down chlorophyll although I forget it's name
Beta-Carotene. Reddish pigment, a type of chlorophyll. The 'big three' pigments are a-chlorophyll, b-Carotene and lutien. The last one is bright yellow!
I dissolved my bho extract in iso. And attempted to wash with activated carbon to remove some chlorophyll. Pick up. This did not seen to help the green at all and then i tried a germicidal uv light this did. I then tried the uv light again after getting flash burn from looking at the light its self hahaha never again. But i tried to get the same results with a more saturated solution of oil and chlorophyll.. its glows pretty red but after 3 days has not broken down the chlorophyll. Why?
Try graphitized carbon, as I have heard it works better; it will also bind some cannabinoids.
Nice article im going to re read it again And again to fully comprehend. Because i attempted this nacl wash 3 month's ago with 1/3 nacl(brine) 1/3 alcohol/ cannabis 1/3 naphtha. To remove a extreme amount of chlorophyll for informative purpose only and was extremely in effective it created even clean separation of all 3 and faild to even slightly remove the chlorophyll.
So I have a solution that is frozen. Do I just take out the ice then let it evaporate, business as usual? Do I have to filter it or anything?
We filter out the waxes that precipitate out of solution and then evaporate away the alcohol.
I grow out doors. I like to let my plants die from frost and let the chlorophyll break down, into carotines. The smoke is smoother. Has anyone played with chemically breaking down chlorophyll in green material to avoid getting it into oil? Over the years I have done alcohol extractions. Now I do butane, but save the material and do an alcohol extraction afterwards, for cooking oil. It would be nice to be able to get rid of the chlorophyll flavor.
So reading further, I find you recommend putting green solutions in sunlight to let the UV do its thing. Thank you for a great, informative site. It is interesting to see the evolution from the 'Isomerizer' system from forty years ago.
We haven't. Anyone else?
Why not just use 7x purity butane in a glass column. It does not carry chlorophyll when done properly. You can use bentonite clay to remove chlorophyll from a hexane solution, but I have never tried with alcohols. Alumina column chromatography works also, but why not just do it right and avoid exogenous chlorophyll from the get-go>?
Avoiding the chlorophyll in the first place, is the best idea, especially if you want monoterpene retention. That is easiest if you use a non polar solvent, but it is water content that causes chlorophyll pickup in the form of micelles, so 7X is little different than utility grade on that issue.
Isomirization most definitely reduces the chlorophyll or I think burns it up when the sulfuric acid refluxes with the oil in your preferred solvent. Post isomirized bho tastes bland and smooth but I'm a fanatic for pure thc without other cannabiniods. If you have ever tried isomirized oil it gives an especially energetic high similer and stronger then equatorial Sativas. Almost aphetamine-ish.
Isomerization works better on material with high CBD, than with recreational strains bred for high THC and low CBD. Even with all the CBD isomerized to THC-8, it adds little if there isn't much there to start with.
PLEASE HELP!!!! I have about 10 lbs of oil that I have extracted using isopropyl that is greener then you could imagine. I need to get the green out or a least most of it. What is your suggestion with such a bulk amount please please help me. Only reason why I use iso is because that is the only solvent I can get my hands on in such quantity (I use about 60 gallons for my process). I have read your article about the hexane salt water wash, only problem is I can not get hexane where I live! Is there another alternative to the hexane? or an alternative to my chlorophyll problem all together. Any suggestions would be so much help.
n-Pentane will also work, and some petroleum ethers, depending on what their MSDS sheet says in in them.
So i dried fresh plant material in a oven at 170 till it would pass through a metal mesh strainer. I achieved a dark dark amber oil from the bho process. It is slightly green and im going to winterize it in to a absolute. I'll expose it to some UV to break down the chlorophyll then filter it later and soforth. I would like to get a blonder oil do you think that using NaCl wash with a iso will achieve a lighter color oil.?
You will get lighter oil if you only lightly break up the material and extract at low temperatures. A brine wash helps remove chlorophyll and water solubles, but it also removes a lot of the lighter terpenes.
Unfortunately I have some green oil I made from toluene after growing bored of Iso extracts. I would like to redissolve it in toluene and clean with 99% iso and saline solution in either a ziplock bag or a twist top gatorade bottle for a make-shift separatory funnel. What're your thoughts on this? All advice cherished and great work, I'm a huge fan respect you all highly for your continuing endeavor
Sounds like it will help. Keep us updated!
Say WHAT>? Toluene in a Ziplock Bag>? Plastic Gatorade Bottle as a Sep Funnel>? I am about bent over laughing. ANYONE worth their salt would NEVER advise using Toluene in plastic. Even if you dont completely dissolve the plastic, you will certainly pick up toxic pthalates and phenols leached directly from the plastic. Toluene is maybe safe with PTFE, but why not just recommend GLASS>??? Your advice, or lack thereof, is dangerous
And where did you see Toulene?
WELL DONE! as a college chemistry major this information is EXACTLY!! what i needed. i love all the articles on the site. understanding the science behind this is so awesome
Great Job All and good explanation Joe.Don't normally respond to posts but you have some great information.I am proud you hail from Oregon.Thank you "old as dirt"K
Would filtering the extract solution (isopropyl alcohol solvent) through high grade activated carbon do any good in removing the chlorophyll? Would this give me a more blonde and more pure end result?
The carbon would remove some of the polar elements like chlorophyll and make a blonder extract. It would make the extract more concentrated and more pure, but will also take away some of the cannabinoids.
How does it make it more concentrated and pure for my target compound if it is taking away precious cannabinoids? I'm assuming it's a neglible amount. Would you consider it worth doing to clean up a QWISO? Thanks again
It removes more chlorophyll than cannabinoids, thus the amount may be less, but what is left is more concentrated. It is certainly worth experimenting with.
Great site, great info and techniques. However... Although I agree with Joe's chemical interactions reasoning and indeed with the technique described for purification, I must add a discrepancy into your presentation of facts. The article states: "Merck Index lists chlorophyll as practically insoluble in non polar solvents, so the difference between practically and totally insoluble may offer a clue, as well as butanes practically insolubility in water." Well I have a quite recent edition of The Merck Index installed on my computer (+ an older physical copy of the book) and I again looked up chlorophyll's spec's only to find that Chlorophyll A and Chlorophyll B (the two forms found in cannabis) are listed as: Chlorophyll A "Freely sol in ether, ethanol, acetone, chloroform, carbon disulfide, benzene. Sparingly sol in cold methanol. Practically insol in petr ether." Chlorphyll B "Sparingly sol in petr ether, ligroin, cold methanol. Freely sol in abs alcohol, ether." (I even copied and pasted these spec's from The Merck Index!) Note that: Chlorophyll A is freely soluble in ether, chloroform, carbon disulfide and benzene which are all non-polar solvents. Chlorphyll B is freely soluble in ether which is a non-polar solvent. However both Chlorophyll's (A&B) are listed in The Merck Index as practically and sparingly soluble respectively in petr ether (also called ligroin just to confuse things), which is the most non-polar of all these solvents. So, to reiterate, although I agree with the reasoning and even methodology given, I have no idea where the statement that "Merck Index lists chlorophyll as practically insoluble in non polar solvents" came from, since both the Tenth and Thirteenth Editions of The Merck Index (which have the same chlorophyll entry anyway) DO NOT LIST EITHER CHLOROPHYLL A OR CHLORPHYLL B AS BEING PRACTICALLY INSOLUBLE IN NON-POLAR SOLVENTS! In fact two forms of chlorophyll (A&B) exist, with different solubility properties, in cannabis and this article lumps them as one (though they do have similar properties). WTF? Sorry for that implied bad language, but you get the drift. I don't think simplification is called for here, since the article purposely goes into an unrestricted technical discussion in order to aid in the production of a purer/better product.
Hexane has a dialect constant of 1.89, diethyl ether is 4.34, methanol is 32.6, acetone 20.7, ethanol 24.6 and though pet ether was not given in the reference (chemfinder) it is generally considered a relatively non polar solvent. This is why it was lumped in with other non polar solvents. Hexane is very non polar relative to ether or acetone so it should only sparingly dissolve chlorophy. The structure that changes chlorophyll A to B is not as relevant as the overall structure that makes chlorophyll a tricky one to get out. Since either will green your product separating them into A and B would be purely academic. The term non polar means compounds containing only C&H and single bonds; the solvents you reference as being non polar contain oxygen, sulfur, chlorine and a rotating set of 3 double bonds which makes them all polar. Hopefully this clarified the generalizations I made in regards to solvents and chlorophyll. Joe
I agree to all the above. I greatly simplified the situation by calling solvents non-polar or polar based upon their general oil/water miscibility characteristics and was indeed contemplating suggesting the partial(?)/degrees-of-polarity of each, but thought that might even further complicate the discussion for many readers (though ironically that was obviously my intention is adding details to the discussion). Yes Sulphur and Oxygen atoms do add degrees of polarity to each solvents molecule and its subsequent properties, so I think maybe we should be concentrating defining the highly specific/unique extraction properties of each solvent under specific conditions - for the wealth & health of all humanity!
Arwhooooo....It was my pleasure dad....hope this keeps the goodies safe.
Way to go Wayward Wolf!!!!!
If chlorophyll pigment was larger (heavier), and the pH is 4, would they settle ? I just bought a qt. 190-proof Ethanol (denatured) and am going to do an extraction, following your instructions. Thank you for this website!
Given enough time and perhaps at low enough temperature, then yes, the chlorophyll should precipitate.
I have done this as an experiment. I added iso to the wet cannabis about half a pint. I pushed on the material for a few sec then filtered using fast paper. I think I did this 2-3 times then placed it in the freezer and waited. It took days for sediment to form for on the bottom. I filtered it with a medium filter. The solution was still green but acceptable. Put in sun in iso . It was better than I expected.